An adrenal incidentaloma (Al) is a supracentimetric adrenal mass detected on imaging performed for another reason. Therefore, it is not discovered during neoplastic staging or exploration of hypertension.
The incidence mirrors the increase in imaging exams and raises three main questions: is this mass secreting? Is it malignant? How can it be managed?
In the vast majority of cases, it appears to be a non-secreting benign adenoma that should be monitored. Hormonal assessment is essential to initiate any specific management and sometimes to guide the diagnostic assessment. Radiological criteria are then used to classify the lesion as potentially benign or malignant.
Adrenal malignancies are rare. Therefore, when they are suspected, management should be multidisciplinary with a surgeon, endocrinologist, oncologist, nuclear radiologist and radiologist, if possible in an expert centre. This requirement has led to the development of national (COMETE (Appendix 1) in France, GANIMED in Germany and NISGAT in Italy) and international (ENSAT in Europe) networks. These networks regularly publish recommendations, facilitate extensive collection of biological tissues and promote the inclusion of patients in therapeutic trials.
The aim of this article is to assist urologists confronted with adrenal incidentaloma, by providing initial oncological management guidance through a malignancy assessment. This is based on the recommendations established by the CCAFU in 2018 [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195] and on the scientific literature available on PubMed in 2020.
Investigation of an Al may reveal lesions of various types that affect the adrenal much more than the medullary cortex. Malignant adrenal tumours (MAT) can be: primary, affecting the cortex of the gland (adrenal cortical carcinoma (ACC) or malignant adrenocortical carcinoma); or its medulla (malignant pheochromocytoma); or secondary to a cancer of another origin (exceptional primary lymphomas, which are mostly bilateral, will not be treated here).
The incidence varies from 3% to more than 10% in adults and increases with age [2Mayo-Smith W.W., Song J.H., Boland G.L., Francis I.R., Israel G.M., Mazzaglia P.J., et al. Management of Incidental Adrenal Masses: A White Paper of the ACR Incidental Findings Committee. J Am Coll Radiol 2017 ; 14 (8) : 1038-1044 [cross-ref], 3Bhargava P., Sangster G., Haque K., Garrett J., Donato M., D'Agostino H. AMultimodality Review of Adrenal Tumors. Curr Probl Diagn Radiol 2019 ; 48 (6) : 605-615 [cross-ref]]. If there is a history of cancer, Als are more common (9-13%) [4Blake M.A., Cronin C.G., Boland G.W. Adrenal imaging. AJR Am J Roentgenol 2010 ; 194 (6) : 1450-1460 [cross-ref]]. Adrenocortical adenomas (75%) and myelolipomas (6%) are benign and are the most common tumours. The probability of malignancy in Al is low (< 5%) [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
|
|
|
Adrenal cortical carcinoma (ACC)
|
This is a rare tumour, derived from the adrenal cortex. The annual incidence is 0.5 to 2 per million inhabitants. ACC is more common in women (55-60%)[1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. It can occur at any age but there is a peak incidence between 40 and 60 years of age [5Fassnacht M., Dekkers O.M., Else T., Baudin E., Berruti A., de Krijger R., et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 (4) : G1-G46]. In most cases, ACC is sporadic and rarely associated with other endocrine neoplasia (NEM type 1, Beckwith-Wiedemann syndrome, Li-Fraumeni syndrome).
Pheochromocytoma (PC) is a rare neuroendocrine tumour derived from chromaffin cells of the adrenal medulla. The annual incidence is 2 to 8 per million adults. A spike in frequency is observed between the ages of 30 and 40. Approximately 10% of pheochromocytomas are malignant [6Harari A. Inabnet 3rd WB. Malignant pheochromocytoma: a review. Am J Surg 2011 ; 201 (5) : 700-708 [inter-ref]]. Size greater than 5 cm, local invasion, internal necrosis, nuclear pleomorphism and hyperchromicity are features suggestive of malignancy, but the only criterion for malignancy is the existence of secondary locations in organs without chromaffin tissue (in order of frequency: lymph nodes, bones, liver, lungs and kidneys) [7Andrade M.O., Cunha V.SD., Oliveira D.C., Moraes O.L., Lofrano-Porto A. What determines mortality in malignant pheochromocytoma? - Report of a case with eighteen-year survival and review of the literature. Arch Endocrinol Metab 2018 ; 62 (2) : 264-269 [cross-ref]].
The majority of malignant PCs (MPCs) are sporadic (75%) but they can also occur in the context of genetic diseases: NEM type 2 (10% of MPCs are associated with multiple endocrine neoplasia syndrome [6Harari A. Inabnet 3rd WB. Malignant pheochromocytoma: a review. Am J Surg 2011 ; 201 (5) : 700-708 [inter-ref]]), von Hippel-Lindau disease, mutation in the succinate dehydrogenase subunit B (SDHB), neurofibromatosis type 1, Sturge-Weber syndrome, and tuberous sclerosis [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. Therefore, in case of MPC, a hereditary context should be systematically ruled out. Oncogenetic consultation is recommended if a genetic disease or bilateral PC is suspected, and in young patients (under 45 years of age) [8Renard J., Clerici T., Licker M., Triponez F. Pheochromocytoma and abdominal paraganglioma. J Visc Surg 2011 ; 148 (6) : e409-16].
In patients with a history of cancer, Al is a metastasis of the previous cancer in 75% of the cases [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]]. The risk of metastasis is estimated to be less than 2% in the absence of any active cancer. If there is a context of cancer and isolated adrenal involvement, the risk of metastasis is approximately 30 to 50%. Conversely, if the patient has no known extra-adrenal cancer, imaging is exceptionally revealing of such primary tumours [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]].
Table recommendation 1Adrenal incidentalomaRecommendationsStrength ratingAls are frequent but rarely malignant. However, a hormonal assessment followed by an aetiological assessment and then a malignancy assessment are recommended for all cases of Al.WeakIf there is a history of extra-adrenal cancer, Al is considered to be metastatic until proven otherwise.WeakThe rarity of adrenal malignancies justifies treatment in an expert centre (and in contact with ENSAT) and inclusion as soon as possible in therapeutic trials.Weak
| Table recommendation 1 - Adrenal incidentaloma |
|
| Recommendations |
Strength rating |
| Als are frequent but rarely malignant. However, a hormonal assessment followed by an aetiological assessment and then a malignancy assessment are recommended for all cases of Al. |
Weak |
| If there is a history of extra-adrenal cancer, Al is considered to be metastatic until proven otherwise. |
Weak |
| The rarity of adrenal malignancies justifies treatment in an expert centre (and in contact with ENSAT) and inclusion as soon as possible in therapeutic trials. |
Weak |
|
The clinical assessment of Al should be clinical and then hormonal, in order to characterise the secretion profile (secretory or non-secretory). The aetiological assessment continues with the repetition or completion of imaging. In case of non-secretory incidentaloma, the fear is that an asymptomatic malignant tumour may develop in the absence of surgery. This is a minor possibility. Identifying indications of malignancy is the third step [10Berland L.L., Silverman S.G., Gore R.M., Mayo-Smith W.W., Megibow A.J., Yee J., et al. Managing incidental findings on abdominal CT: white paper of the ACR incidental findings committee. J Am Coll Radiol 2010 ; 7 (10) : 754-773 [cross-ref]].
Subclinical (âpre-Cushingâ) hypercortisolism or undetected pheochromocytoma should be investigated. The other abnormal secretions (virilising or feminising tumour) are rarely asymptomatic and are hardly ever noted alongside adrenal incidentaloma [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]].
|
| Interview and clinical examination |
The examination should identify signs of subclinical hormone hypersecretion (history, benign symptoms) (Table 1). It may also reveal the potential consequences of an abdominal mass syndrome or signs suggestive of malignancy, (especially when they are recent and of rapid onset): deep lumbago, fever of unknown origin, anorexia +/â' weight loss.
High blood pressure (HTN) and specific signs of MAT (lumbar involvement) are ruled out by the clinical examination.
|
| Special clinical features |
The clinical picture of PC (due to excess catecholamines) may include: hypertension (HTN) with hypokalemia, weight loss and pallor due to peripheral vasoconstriction. The typical symptomatic triad known as the « Menard triad » (pulsatile headaches, heart palpitations and tachycardia and profuse sweating) is inconsistent.
None of these clinical signs point to PC malignancy [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. The persistence of clinical signs after adrenalectomy for PC is suggestive of residual metastases and therefore of MPC [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
MPCs are secretory (catecholamines) in 85% of cases. If that is the case, 3 types of symptomatic complications can occur: cardiovascular disorders (hypertension, dilated cardiomyopathy, etc.), gastrointestinal disorders (severe constipation) and bone events (70% of the patients develop bone metastases, mostly lytic, which in 80% of the cases lead to pain, fractures or spinal cord compression) [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22].
|
| Adrenal cortical carcinoma |
ACCs, which are sometimes asymptomatic, are secretory in 50 to 60% of the cases: Cortisol with Cushing's syndrome (â 30%:), androgens with signs of virilisation in women (â 20%), oestrogens with signs of feminisation in men (â 10%), mixed secretion (â 35%).
One third of virilizing tumours are malignant. Feminising tumours are almost always malignant and account for 10% of ACCs [12Song J.H., Chaudhry F.S., Mayo-Smith W.W. The incidental adrenal mass on CT: prevalence of adrenal disease in 1,049 consecutive adrenal masses in patients with no known malignancy. AJR Am J Roentgenol 2008 ; 190 (5) : 1163-1168 [cross-ref]].
In case of synchronous AM, the clinical signs are dominated by those of the primary cancer and possible of other locations. While AM is metachronous and isolated, it is often asymptomatic and is discovered during surveillance of the primary cancer [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Before re-evaluating imaging criteria, a minimum biology assessment should be performed.
In addition to fasting blood glucose (diabetes possible in hypercortisolism and PC), hormone assays (Table 2) are recommended during an endocrinology consultation (directed questioning, age, co-morbidities).
Diagnostic guidelines can be established by the hormone profile.
The secretion of sex or mixed hormones (Cortisol and sex hormones) is an indication of malignancy in adrenal cortical tumour.
Plasma methoxytyramine is a predictive marker of PC malignancy [13Berruti A., Baudin E., Gelderblom H., Haak H.R., Porpiglia F., Fassnacht M., et al. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up Ann Oncol 2012 ; 23 : vii131-8, 14Eisenhofer G., Lenders J.W., Siegert G., Bornstein S.R., Friberg P., Milosevic D., et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status Eur J Cancer 2012 ; 48 (11) : 1739-1749 [cross-ref]]. One tool that may be useful in differentiating a benign from a malignant adrenal cortical tumour is the urine steroid profile measured by mass spectrometry (GC-MS or LC-MS) [15Kerkhofs T.M., Kerstens M.N., Kema I.P., Willems T.P., Haak H.R. Diagnostic Value of Urinary Steroid Profiling in the Evaluation of Adrenal Tumors. Horm Cancer 2015 ; 6 (4) : 168-175 [cross-ref]].
|
| Complementary biological assessment |
Genetic testing is only performed in a context of hereditary disease (family history, young patients, bilateral tumours, extra-adrenal location, MPC, ACC).
The size of an Al can be predictive of its malignancy regardless of the imaging modality. However, a recent study seems to lower the predictive value of ACC based solely on the criterion of size [17Kahramangil B, Kose E, Remer EM, Reynolds JP, Stein R, Rini B, et al. A Modern Assessment of Cancer Risk in Adrenal Incidentalomas: Analysis of 2219 Patients. Ann Surg 2020.].
Above 6 cm, the proportion of malignant tumours is 25%, while it is less than 2% for masses of less than 4 cm [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref],18Vural V., Kilinc E.M., Saridemir D., Gok I.B., Huseynov A., Akbarov A., et al. Association between Tumor Size and Malignancy Risk in Hormonally Inactive Adrenal Incidentalomas. Cureus 2020 ; 12 (1) : e6574]. Therefore, a tumour diameter > 6 cm is always an argument for malignancy.
Considering the generally rapid tumour growth of MAT, it has been suggested that for monitored incidentalomas, control CT scans should be performed at 6 months and 1 year. In case of stability and a non-secretory Al, there is no indication for routine radiological follow-up, [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195,19Sebe P., Rigaud J., Avances C., Brunaud L., Caillard C., Camparo P., et al. [CCAFU's contribution to the French National Cancer Institute's reference frame: Adrenal malignant tumors] Prog Urol 2013 ; 23 : S167-74] but the appearance of clinical symptoms or signs should be monitored.
For ACCs, the specificity is 52, 80, 95 and 98% respectively for diameters > 4 cm, > 6 cm, > 8 cm or > 10 cm [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
For MPCs, the specificity is 20, 65 and 89% respectively for diameters of 4 cm, 6 cm and 8 cm [20Shen W.T., Sturgeon C., Clark O.H., Duh Q.Y., Kebebew E. Should pheochromocytoma size influence surgical approach? A comparison of 90 malignant and 60 benign pheochromocytomas Surgery 2004 ; 136 (6) : 1129-1137 [cross-ref]].
|
| Characteristics of computed tomography (CT scan) |
The examination of spontaneous density differentiates adenomas, rich in microscopic fat, from malignant lesions, which are poorer, with a sensitivity and specificity of 71 and 98%, respectively [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]]. Therefore, malignant tumours could have a higher spontaneous density than benign tumours. In the series by Szolar et al, the mean densities of adrenal tumours were: 39 HU for ACCs, 44 HU for MPCs and 34 HU for adrenal metastases (compared to 8 HU for adrenal adenomas). This trend could be confirmed on the densities 10 minutes after injection [21Szolar D.H., Korobkin M., Reittner P., Berghold A., Bauernhofer T., Trummer H., et al. Adrenocortical carcinomas and adrenal pheochromocytomas: mass and enhancement loss evaluation at delayed contrast-enhanced CT. Radiology 2005 ; 234 (2) : 479-485 [cross-ref]].
The analysis of mass enhancement after injection also helps in characterisation. Adenomas have a greater absolute (taking into account spontaneous density) or relative washout than metastases (excluding hypervascular metastases of clear cell renal cell carcinoma). The washout calculation does not differentiate an adenoma from pheochromocytoma, which is a hypervascular tumour.
Kahramangil confirms that the suspicion of Al malignancy is the result of a combination of clinical, hormonal and imaging evaluations [17Kahramangil B, Kose E, Remer EM, Reynolds JP, Stein R, Rini B, et al. A Modern Assessment of Cancer Risk in Adrenal Incidentalomas: Analysis of 2219 Patients. Ann Surg 2020.].
|
| Characteristics of magnetic resonance imaging (MRI) |
Abdominal MRI provides no additional diagnostic information compared to CT (the sensitivity and specificity are lower): 78 and 87% respectively in tissue characterisation [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]]). Contrast uptake after gadolinium injection is important with a hyper signal evocative but not specific to MPC on T2 sequences. The very slow washout after gadolinium injection is suggestive of ACC. In T1 and T2, benign tissue lesions have a signal intensity equal to or slightly less than that of the normal liver [23Wu D., Tischler A.S., Lloyd R.V., DeLellis R.A., de Krijger R., van Nederveen F., et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol 2009 ; 33 (4) : 599-608 [cross-ref]]. Pheochromocytoma has a hyper T2 signal. Haemorrhagic changes may alter the signal: adrenal to liver ratio > 3 and a rapid and intense enhancement with gadolinium injection.
In classical sequences, MRI would provide a better assessment of local and venous invasion of a potentially malignant tumour, due to better contrast resolution, but the spatial resolution of the CT scan is better. Therefore, in theory it can supplement abdominal CT data to refine the staging of locoregional, metastatic, vascular or lymph node spread. Even in these indications, current improvements in CT scans limit the use of MRI for many indications. On the other hand, it is a surveillance tool for young patients, to limit repeated irradiation.
The phase-opposed phase sequence or magnetic resonance spectroscopy is useful. A decrease in the signal in the opposed phase (water + fat / fat - water) of 2O% would indicate an adenoma [24Gaujoux S., Mihai R. joint working group of E, Ensat. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma Br J Surg 2017 ; 104 (4) : 358-376 [cross-ref]], usually rich in microscopic fat (high specificity and sensitivity) [25Platzek I., Sieron D., Plodeck V., Borkowetz A., Laniado M., Hoffmann R.T. Chemical shift imaging for evaluation of adrenal masses: a systematic review and meta-analysis. Eur Radiol 2019 ; 29 (2) : 806-817 [cross-ref]].
Table recommendation 2Radiological assessment of adrenal tumoursRecommendationsStrengh ratingThe risk of malignancy in AT increases with the size of the lesion.StrongDuring Ml surveillance: rapid growth of an AT is cause for suspicion of malignancy.StrongAn Ml with a normal hormone balance and a CT scan showing a density < 10 HU, with homogeneous parenchyma and rapid washout is considered a benign adenoma. When it is less than 4 cm, no paraclinical follow-up is necessary [16].StrongA non-secreting Ml of > 4 cm, without any negative criteria on the non-injected CT scan may: justify a complementary imaging test, be monitored by CT or MRI within a year, or be resected immediately [16].StrongOn CT: a spontaneous density > 20 HU, a heterogeneous aspect, hypervascularisation, reduced wash-out and/or irregular borders (local invasion) are indicative of a risk of AT malignancy.StrongMRI is a second-line examination in AT:- Ml with contraindication to CT scan with injection.- Atypical AT on CT scan (density > 10 HU): a signal drop in phase opposition is a sign of a benign tumour [26].- AT suspected of being malignant (to specify local and remote invasion).StrongKey: AT = Adrenal tumour, MI = Malignant Incidentaloma; HU = Hounsfield Units.
| Table recommendation 2 - Radiological assessment of adrenal tumours |
|
| Recommendations |
Strengh rating |
| The risk of malignancy in AT increases with the size of the lesion. |
Strong |
| During Ml surveillance: rapid growth of an AT is cause for suspicion of malignancy. |
Strong |
| An Ml with a normal hormone balance and a CT scan showing a density < 10 HU, with homogeneous parenchyma and rapid washout is considered a benign adenoma. When it is less than 4 cm, no paraclinical follow-up is necessary [16Fassnacht M., Arlt W., Bancos I., Dralle H., Newell-Price J., Sahdev A., et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016 ; 175 (2) : G1-G34]. |
Strong |
| A non-secreting Ml of > 4 cm, without any negative criteria on the non-injected CT scan may: justify a complementary imaging test, be monitored by CT or MRI within a year, or be resected immediately [16Fassnacht M., Arlt W., Bancos I., Dralle H., Newell-Price J., Sahdev A., et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016 ; 175 (2) : G1-G34]. |
Strong |
| On CT: a spontaneous density > 20 HU, a heterogeneous aspect, hypervascularisation, reduced wash-out and/or irregular borders (local invasion) are indicative of a risk of AT malignancy. |
Strong |
| MRI is a second-line examination in AT:
- Ml with contraindication to CT scan with injection.
- Atypical AT on CT scan (density > 10 HU): a signal drop in phase opposition is a sign of a benign tumour [26].- AT suspected of being malignant (to specify local and remote invasion). |
Strong |
|
| Key: AT = Adrenal tumour, MI = Malignant Incidentaloma; HU = Hounsfield Units. |
|
|
| At the end of this assessment |
Kahramangil confirms that the suspicion of Al malignancy is the result of a combination of clinical, hormonal explorations and imaging characteristics [17Kahramangil B, Kose E, Remer EM, Reynolds JP, Stein R, Rini B, et al. A Modern Assessment of Cancer Risk in Adrenal Incidentalomas: Analysis of 2219 Patients. Ann Surg 2020.]. Certain diagnostic hypotheses may also be evoked, which could justify a scintigraphic (functional) exploration to complete the assessment and/or confirm the diagnosis.
|
| Characteristics of scintigraphy |
|
| Positron emission tomography (PET) with fluorodeoxyglucose (18F) or 2-deoxy-2-(18F) fluoro-D-glucose (18F-FDG) |
When there is a hypermetabolic adrenal lesion with 18F-FDG, there are four main diagnoses: PC, ACC, AM or lymphoma.
If ACC is suspected, 18FDG PET is the reference scintigraphic examination, both at the diagnostic phase and for follow-up [24Gaujoux S., Mihai R. joint working group of E, Ensat. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma Br J Surg 2017 ; 104 (4) : 358-376 [cross-ref],27Libe R. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Front Cell Dev Biol 2015 ; 3 : 45]. PET is also useful in the staging of ACC for the diagnosis of distant metastases.
This test is used to calculate the ratio of the SUVmax (maximal Standard Uptake Value ) of the tumour to that of the liver (SUVmax tumour/SUVmax liver). A ratio ⥠1.45 is highly predictive of malignancy [28Thompson L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002 ; 26 (5) : 551-566 [cross-ref]].
18F-FDG PET seems to have a different value for characterising tumours of the medulla, as it is known that both benign and malignant PCs can show uptake of 18F-FDG [29Tenenbaum F., Lataud M., Groussin L. [Update in adrenal imaging] Presse Med 2014 ; 43 : 410-419 [inter-ref]]. It is now recommended preoperatively for staging malignant pheochromocytomas [30Plouin P.F., Amar L., Dekkers O.M., Fassnacht M., Gimenez-Roqueplo A.P., Lenders J.W., et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016 ; 174 (5) : G1-G10]. It is also now authorised in France when an adrenal incidentaloma has been identified [31Taïeb D., Salaün P.Y. Recommandations de bonne pratique clinique pour l'utilisation de la TEP en cancérologie Incidentalomes surrenaliens. Médecine Nucléaire 2019 ; 43 : 66-68].
|
| Positron emission tomography with 18F-DOPA (Fluoro- 18-L-Dihydroxyphenylalanine) |
If PC is suspected, the tracer of choice is 18F-DOPA. It is useful for a positive diagnosis and the identification of possible secondary locations, with sensitivity close to 100%. It is more sensitive than MIBG and can be coupled with 18-FDG [32Taïeb D., Timmers H.J., Hindie E., Guillet B.A., Neumann H.P., Walz M.K., et al. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2012 ; 39 (12) : 1977-1995].
|
| lodine-123 meta-iodo-benzylguanidine (123l-MIBG) scintigraphy |
The fixation of 123I-MIBG, which is a specific adrenal medulla tracer, to an adrenal mass, is very indicative of PC [29Tenenbaum F., Lataud M., Groussin L. [Update in adrenal imaging] Presse Med 2014 ; 43 : 410-419 [inter-ref]]. Classically used since the 1980s to confirm the diagnosis of PC, it can be useful when methoxylated derivatives are limited or variable from one sample to another. When PC is diagnosed, it also eliminates other locations or rare metastases [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]] but for that purpose it is gradually being replaced by PET (it is only indicated when PET is unavailable).
In case of PCM, its use should be limited to patients who can respond to internal radiotherapy (or metabolic radiotherapy) with iodine 131 (131l), vectorised to MIBG (predictive of the effectiveness of this metabolic radiotherapy) [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22,33Lenders J.W., Duh Q.Y., Eisenhofer G., Gimenez-Roqueplo A.P., Grebe S.K., Murad M.H., et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014 ; 99 (6) : 1915-1942 [cross-ref]].
Table recommendation 3Functional imaging of adrenal incidentalomaAssessment of Al by functional imagingStrengh rating18F-FDG PET scan is systematically recommended in case of suspected ACC and Al with a history of extra-adrenal cancer.WeakWhen MPC is suspected, an 18F-FDG PET scan is recommended and can be coupled with 18F-DOPA.StrongThe use of 123I-MIBG scintigraphy in MPC is limited to candidates for metabolic radiotherapy (131l vectorised by this marker).Weak
| Table recommendation 3 - Functional imaging of adrenal incidentaloma |
|
| Assessment of Al by functional imaging |
Strengh rating |
| 18F-FDG PET scan is systematically recommended in case of suspected ACC and Al with a history of extra-adrenal cancer. |
Weak |
| When MPC is suspected, an 18F-FDG PET scan is recommended and can be coupled with 18F-DOPA. |
Strong |
| The use of 123I-MIBG scintigraphy in MPC is limited to candidates for metabolic radiotherapy (131l vectorised by this marker). |
Weak |
|
|
|
|
The place of percutaneous biopsy
|
Percutaneous biopsy has a very limited role. It is not recommended in patients with no history of neoplastic disease [34Tabarin A., Bardet S., Bertherat J., Dupas B., Chabre O., Hamoir E., et al. Exploration and management of adrenal incidentalomas. French Society of Endocrinology Consensus. Ann Endocrinol (Paris) 2008 ; 69 (6) : 487-500 [inter-ref]]. The only indication is a suspicion of adrenal metastasis or of retroperitoneal lymphoma or sarcoma [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Even in these contexts, a subclinical PC should first be formally ruled out because the prevalence of subclinical PC in patients with extra-adrenal cancer is relatively high, ranging from 5% to 25% [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]]. Exceptionally, it may be necessary (after ruling out PC) to confirm a diagnosis of adrenal lesion that is immediately metastatic and unresectable (in which case it should be associated with anti-SF1 immunohistochemistry labelling). If MPC is suspected, traditionally, it is contraindicated (risk of malignant hypertension due to catecholamines overflow) [16Fassnacht M., Arlt W., Bancos I., Dralle H., Newell-Price J., Sahdev A., et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016 ; 175 (2) : G1-G34].
If ACC is suspected, it is contraindicated [24Gaujoux S., Mihai R. joint working group of E, Ensat. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma Br J Surg 2017 ; 104 (4) : 358-376 [cross-ref]] because of the risk of tumour dissemination due to capsular rupture [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. However, it may be of interest in case of uncertainty about whether it is an adrenal metastasis or ACC if it is certain that it is non-secretory [35Fassnacht M., Libe R., Kroiss M., Allolio B. Adrenocortical carcinoma: a clinician's update. Nat Rev Endocrinol 2011 ; 7 (6) : 323-335 [cross-ref]].
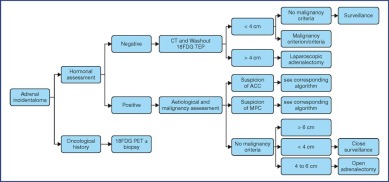
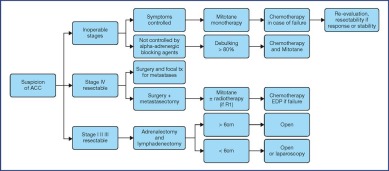
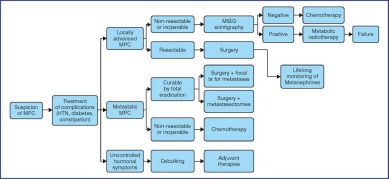
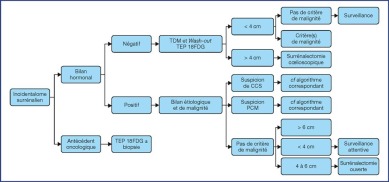
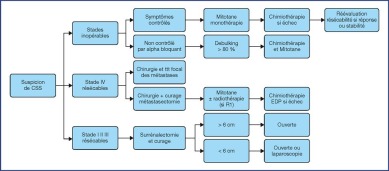
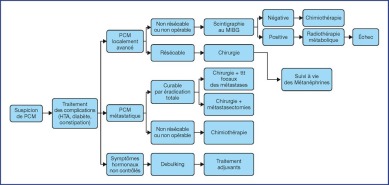
The treatment recommendations are presented as decision algorithms, taken from the CCAFU recommendations published in 2018 [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. The first proposes management of Al based on the results of the malignancy assessment (Figure 1). Specific algorithms are proposed for suspected ACC (Figure 2) and suspected MPC (Figure 3).
Figure 1.
Decisional algorithm for Al [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
(Abbreviations: 18FDG PET = Fluorodeoxyglucose (18F) Positron Emission Tomography; ACC = Adrenal Cortical Carcinoma; MPC = Malignant Pheochromocytoma)
Figure 2.
Decisional algorithm for ACC [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
(Abbreviations: tx = treatment; R0 = no residual tumour; R1 = microscopic residual tumour)
Figure 3.
Decisional algorithm for MPC [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
(Abbreviations: MPC = malignant pheochromocytoma; HTN = hypertension; tx = treatment)
|
|
|
Postoperative oncological assessment
|
|
|
|
Adrenal cortical carcinoma
|
|
| WEISS histopathological prognostic score |
In case of a doubtful diagnosis of ACC, the slides should be reread by a specialised pathologist, within the COMETE network. Double reading is recommended within the network.
If there is any doubt as to whether or not the tumour is of cortical origin, an immunohistochemical examination of Steroidogenic Factor-1 (SF-1 ) expression should be systematically performed because it is the most sensitive and specific adrenal cortex marker [36Sbiera S., Schmull S., Assie G., Voelker H.U., Kraus L., Beyer M., et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J Clin Endocrinol Metab 2010 ; 95 (10) : E161-71].
In established adrenal cortical carcinoma, the pathological examination is crucial to establish the diagnosis. It allows definitive establishment of the pTNM stage, the resection status « R » and especially the calculation of the Weiss histoprognostic score (Table 3). Therefore, malignancy is diagnosed when a localised tumour has a Weiss score > 3 and/or in the event of local invasion or distant metastases [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195,5Fassnacht M., Dekkers O.M., Else T., Baudin E., Berruti A., de Krijger R., et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 (4) : G1-G46,26Libe R., Assie G. [Adrenocortical carcinoma: Update in 2014] Presse Med 2014 ; 43 : 401-409 [cross-ref], 27Libe R. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Front Cell Dev Biol 2015 ; 3 : 45,37Baudin E. Endocrine Tumor Board of Gustave R. Adrenocortical carcinoma. Endocrinol Metab Clin North Am 2015 ; 44 (2) : 411-434 [inter-ref]].
|
| TNM classification of ACCs |
The 8th edition of the TNM (2016) does not have an update from the 2009 version (Table 4) [38Fassnacht M., Johanssen S., Quinkler M., Bucsky P., Willenberg H.S., Beuschlein F., et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer 2009 ; 115 (2) : 243-250 [cross-ref], 39Amin M.B., Greene F.L., Edge S.B., Compton C.C., Gershenwald J.E., Brookland R.K., et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more âpersonalizedâ approach to cancer staging. CA Cancer J Clin 2017 ; 67 (2) : 93-99 [cross-ref]]. It has a prognostic value and is useful for therapeutic management.
|
|
|
Malignant pheochromocytoma
|
In general, the histological diagnosis of PC is not problematic, but the criteria for malignancy are debated [40Patey M. [Pheochromocytoma and the diagnosis of malignancy: recent data and the role of the pathologist] Ann Pathol 2008 ; 28 (1) : S42-4 [inter-ref]]. Capsular invasion and vascular invasion are considered high-risk criteria for malignancy but are not always associated with a metastatic disease [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195,22Ng C.S., Altinmakas E., Wei W., Ghosh P., Li X., Grubbs E.G., et al. Utility of Intermediate-Delay Washout CT Images for Differentiation of Malignant and Benign Adrenal Lesions: A Multivariate Analysis. AJR Am J Roentgenol 2018 ; 211 (2) : W109-W115 [inter-ref]].
At present, the only formal proof of malignancy is the invasion of neighbouring organs or distant metastases [6Harari A. Inabnet 3rd WB. Malignant pheochromocytoma: a review. Am J Surg 2011 ; 201 (5) : 700-708 [inter-ref],23Wu D., Tischler A.S., Lloyd R.V., DeLellis R.A., de Krijger R., van Nederveen F., et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol 2009 ; 33 (4) : 599-608 [cross-ref],40Patey M. [Pheochromocytoma and the diagnosis of malignancy: recent data and the role of the pathologist] Ann Pathol 2008 ; 28 (1) : S42-4 [inter-ref]].
Although not validated (and sometimes inconsistent with other immunohistochemical criteria), the most widely used prognostic score is the Pheochromocytoma of the Adrenal Gland Scaled Score (PASS), proposed in 2002 by Thompson [28Thompson L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002 ; 26 (5) : 551-566 [cross-ref]]. The score is based on the items identified in Table 5. A score of less than 4 is in favour of benignity and more than 6 is in favour of malignancy. It is not recommended in common practice [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Primary cancers are mainly lung cancer (35%), kidney cancer, breast cancer, malignant melanoma, stomach cancer, colorectal cancer and lymphoma. An isolated adrenal location is rare, but they often remain confined to the gland, hence the interest of surgical excision [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Adrenal metastases are classified as M+ in the TNM classification of primary cancer.
Table recommendation 4Recommendation for the histology of adrenal tumoursRecommendationsStrengh ratingThe definitive diagnosis of ACC is histological, which allows the TNM classification to be established.StrongFor PC: no histological criteria are specific to its malignant character, besides the invasion of neighbouring organs.StrongFor PC: capsular invasion and vascular invasion are inconsistent risk factors for malignancy. The PASS score is imprecise and is therefore not recommended for use in routine practice.WeakThere is no TNM classification for MPC.Strong
| Table recommendation 4 - Recommendation for the histology of adrenal tumours |
|
| Recommendations |
Strengh rating |
| The definitive diagnosis of ACC is histological, which allows the TNM classification to be established. |
Strong |
| For PC: no histological criteria are specific to its malignant character, besides the invasion of neighbouring organs. |
Strong |
| For PC: capsular invasion and vascular invasion are inconsistent risk factors for malignancy. The PASS score is imprecise and is therefore not recommended for use in routine practice. |
Weak |
| There is no TNM classification for MPC. |
Strong |
|
|
|
|
Adrenal cortical carcinoma
|
Advanced age and Cortisol secretion appear to be associated with a bleaker prognosis [26Libe R., Assie G. [Adrenocortical carcinoma: Update in 2014] Presse Med 2014 ; 43 : 401-409 [cross-ref]].
Several molecular markers could also be associated with a poor prognosis. They are measured by immunohistochemistry (nuclear accumulation of p53, high intensity of SF-1 labelling, nuclear accumulation of beta-catenin), or by molecular biology techniques (gene expression profile in favour of a tumour with a poor prognosis, high level of methylation of the promoter region of genes, a particular combination of chromosomal losses and gains) [26Libe R., Assie G. [Adrenocortical carcinoma: Update in 2014] Presse Med 2014 ; 43 : 401-409 [cross-ref]], but their use is not recommended in current practice.
The proportion of cells in mitosis (grade) is a recognised criterion of aggressiveness in histology. In ACC, the recognised histological prognostic criterion is the grade expressed by analysis of an anti-Ki 67 immunohistochemical marker. The thresholds that define a negative prognosis are: > 10%: High risk; > 30%: very high risk. Proliferation indices, assessment of the quality of the resection, and lymph node status also provide information on the prognosis.
|
| Prognostic classifications |
The European Network for the Study of Adrenal Tumours (ENS@T) classification [38Fassnacht M., Johanssen S., Quinkler M., Bucsky P., Willenberg H.S., Beuschlein F., et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer 2009 ; 115 (2) : 243-250 [cross-ref]] is superimposed on the AJCC classification, which groups ACCs into 4 prognostic groups by incorporating the TNM (Table 6) [39Amin M.B., Greene F.L., Edge S.B., Compton C.C., Gershenwald J.E., Brookland R.K., et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more âpersonalizedâ approach to cancer staging. CA Cancer J Clin 2017 ; 67 (2) : 93-99 [cross-ref]].
Survival at 5 years is different at these stages: 66-82% for stage 1, 58-64% for stage 2, 24-50% for stage 3 and 0-17% for stage 4 [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195,13Berruti A., Baudin E., Gelderblom H., Haak H.R., Porpiglia F., Fassnacht M., et al. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up Ann Oncol 2012 ; 23 : vii131-8].
Resection at healthy margins is a prerequisite for long-term survival [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. Similarly, capsular rupture is predictive of recurrence.
A group of American experts, United States ACC Study Group, has proposed changing the TNM classification to include lymphovascular invasion, which would improve the prognostic value primarily in the T2 and T3 stages [41Crona J., Beuschlein F., Pacak K., Skogseid B. Advances in adrenal tumors 2018. Endocr Relat Cancer 2018 ; 25 (7) : R405-R420, 42Poorman C.E., Ethun C.G., Postlewait L.M., Tran T.B., Prescott J.D., Pawlik T.M., et al. A Novel T-Stage Classification System for Adrenocortical Carcinoma: Proposal from the US Adrenocortical Carcinoma Study Group. Ann Surg Oncol 2018 ; 25 (2) : 520-527 [cross-ref]].
|
|
|
Malignant pheochromocytoma
|
No validated prognostic factors exist for MPC. The 5-year survival rate ranges from 40 to 77% [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22].
Tumour growth is the main cause of death in MPC. Therefore, tumour control should be the primary objective of MPC management. However, clinical manifestations due to excess catecholamines (high blood pressure, constipation, etc.) should be treated because they might be responsible for 30% of the deaths from MPC [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22]. Finally, death from another cancer is possible, especially in the context of genetic diseases (MEN) [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22,43Ayala-Ramirez M., Feng L., Habra M.A., Rich T., Dickson P.V., Perrier N., et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer 2012 ; 118 (11) : 2804-2812 [cross-ref]].
However, several negative factors have been suggested for MPC: large tumour volume, the existence or number of visceral metastases and the presence of a mutation in the SDHB (Succinate dehydrogenase B) gene [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22].
Table recommendation 5Recommendations for the prognosis of adrenal tumoursRecommendationsStrengh ratingHigh grade ACCs (>20 mitoses/50ch) and/or immunohistochemical expression of Ki-67 are indications of malignancy associated with a poor prognosis.WeakThe pathology report for ACC should specify at least: the Weiss score, T-stage, N-status, margin quality (R) and exact % Ki67 expressionStrongFor ACC, the ENSAT classification (from the TNM) is recommended and is of major interest because it has prognostic value and guides treatment.StrongThere are no validated prognostic factors in MPC. However some factors are negative (large tumour volume, visceral metastases, SDHB mutation).Weak
| Table recommendation 5 - Recommendations for the prognosis of adrenal tumours |
|
| Recommendations |
Strengh rating |
| High grade ACCs (>20 mitoses/50ch) and/or immunohistochemical expression of Ki-67 are indications of malignancy associated with a poor prognosis. |
Weak |
| The pathology report for ACC should specify at least: the Weiss score, T-stage, N-status, margin quality (R) and exact % Ki67 expression |
Strong |
| For ACC, the ENSAT classification (from the TNM) is recommended and is of major interest because it has prognostic value and guides treatment. |
Strong |
| There are no validated prognostic factors in MPC. However some factors are negative (large tumour volume, visceral metastases, SDHB mutation). |
Weak |
|
|
|
|
Adrenal cortical carcinoma
|
After complete resection, clinical, hormonal and imaging surveillance (thoraco-abdomino-pelvic CT scan or 18FDG-PET) every 3 months for 2 years, then every 3 to 6 months for 3 years is recommended [5Fassnacht M., Dekkers O.M., Else T., Baudin E., Berruti A., de Krijger R., et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 (4) : G1-G46]. Beyond 5 years, surveillance is moderate but should be considered on a case by case basis.
For advanced ACCs, the surveillance protocol depends on prognostic factors, expected treatment efficacy and treatment-related toxicity, as well as available alternative treatment options.
A regular hormonal assessment is recommended for all patients [5Fassnacht M., Dekkers O.M., Else T., Baudin E., Berruti A., de Krijger R., et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 (4) : G1-G46].
|
|
|
Malignant pheochromocytoma
|
Monitoring MPC is based on plasma or urine metanephrine testing [33Lenders J.W., Duh Q.Y., Eisenhofer G., Gimenez-Roqueplo A.P., Grebe S.K., Murad M.H., et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014 ; 99 (6) : 1915-1942 [cross-ref]]. A half-yearly frequency for the first year and then at least annually for 5 years is recommended. Recurrence may occur very late and a genetic diathesis may remain undiagnosed, suggesting the need for lifelong surveillance [8Renard J., Clerici T., Licker M., Triponez F. Pheochromocytoma and abdominal paraganglioma. J Visc Surg 2011 ; 148 (6) : e409-16]. Surveillance can be spaced out over time (every 6 months after 10 years) [6Harari A. Inabnet 3rd WB. Malignant pheochromocytoma: a review. Am J Surg 2011 ; 201 (5) : 700-708 [inter-ref]]. An 18 FDG +/- DOPA PET is recommended in case of doubt.
The eradication of AM does not influence the specific follow-up for primary cancer. In case of conservative treatment, 18FDG-PET imaging may complement the usual follow-up.
Als are frequent but rarely malignant. Therefore, the highly specific treatment of MATs justifies their inclusion in the COMETE network in France (Appendix 1). A minimal endocrine assessment is necessary even if metastasis is suspected. Management varies according to the diagnostic orientation. As for any rare disease, management in an expert centre is recommended.
|
|
|
Appendix 1. List of « COMETE » reference centres - Adrenal cancers
|
|
• |
National Expert Reference Centre (co-coordination): Paris - Villejuif and associated centres (Ile de France Region) |
Heads: Prof. BERTHERAT (Cochin), Dr. Baudin (Institut Gustave Roussy)
Associated centres in Ile de France (contacts): HEGP (Dr. Amar), St Antoine (Dr. Donadille), Ambroise Paré (Prof. Raffin-Sanson), Pitié Salpetrière (Dr. Ghander), Saint Louis (Dr. Chougnet), Reims (Prof. Delemer)
|
• |
9 Regional Expert Centres - (Associated centres) |
|
â |
Competence Centre for Adrenal Cancer - Pays de Loire Region: Angers |
Head: Prof. Rohmer, Angers University Hospital Centre (CHU)
Associated centres (contacts): Brest (Prof. Kerlan), Tours (Dr. Pierre), Nantes (Dr. Drui)
|
â |
Competence Centre for Adrenal Cancer- Bourgogne Region: Dijon |
Heads: Prof. Vergès and Dr. Zanetta, Dijon Burgundy CHU - François Mitterrand Hospital
Associated centre (contact): Besançon (Dr. Schillo)
|
â |
Competence Centre for Adrenal Cancer - Rhône-Alpes Region: Grenoble |
Head: Prof. Chabre, Grenoble CHU, North site - Albert Michallon Hospital
Associated centres (contacts): Clermont-Ferrand (Prof. Tauveron), Lyon (Prof. Borson-Chazot)
|
â |
Competence Centre for Adrenal Cancer - Nord-Pas-de-Calais Region: Lille |
Head: Prof. Vantyghem, Lille Regional University Hospital Centre (CHRU) - Claude Huriez Hospital
Associated centre (contact): Amiens (Prof. Desailloud)
|
â |
Competence Centre for Adrenal Cancer- PACA Region: Marseille |
Head: Prof. Niccoli, Marseille CHU - La Timone Hospital
Associated centres (contacts): Marseille (Prof. Castinetti), Montpellier (Dr. Raingeard), Nice (Prof. Sadoul)
|
â |
Competence Centre for Adrenal Cancer- Aquitaine Region: Bordeaux |
Head: Prof. Tabarin, Bordeaux - Southern Hospital Group (GH Sud) - Haut-Lévêque Hospital
Assodated centres (contacts): Limoges (Prof. Archambeaud), Poitiers (Prof. Marechaud), Pointe-à-Pitre (Dr. Cephise), Papeete (Dr. Rachedi), Reunion Island (Dr. Lemoullec)
|
â |
Competence Centre for Adrenal Cancer, Upper Normandy Region: Rouen |
Head: Prof. Lefebvre, Rouen CHU
Associated centre (contact): Caen (Prof. Reznik)
|
â |
Competence Centre for Adrenal Cancer- Alsace Region: Strasbourg |
Head: Prof. Goichot, Strasbourg CHU - Hautepierre Hospital
Associated centre (contact): Nancy (Prof. Brunaud)
|
â |
Competence Centre for Adrenal Cancer - Midi-Pyrenées Region: TOULOUSE |
Head: Prof. Caron, Toulouse CHU - Larrey Hospital No associated centre
Un incidentalome surrénalien (IS) est une masse surréna-lienne, supracentimétrique, découverte fortuitement lors d'un examen radiologique réalisé pour une autre indication. Cette définition exclut sa découverte au cours d'un bilan d'extension néoplasique ou de l'exploration d'une hypertension artérielle (HTA).
L'incidence suit l'augmentation des examens d'imagerie et soulève trois questions principales : cette masse est-elle sécrétante ? Est-elle maligne ? Comment la prendre en charge ?
Dans la grande majorité des cas, il s'agira d'un adénome bénin non sécrétant, à surveiller. Le bilan hormonal sera indispensable pour déclencher une éventuelle prise en charge spécifique et parfois pour orienter le bilan diagnostique. Des critères radiologiques permettent alors de classer la lésion en potentiellement bénigne ou maligne.
Les tumeurs malignes surrénaliennes étant rares, leur suspicion justifie une prise en charge multidisciplinaire associant chirurgien, endocrinologue, oncologue, médecin nucléaire et radiologue, si possible dans un centre expert. Cette exigence a conduit au développement de réseaux nationaux (COMETE [Annexe 1] en France, GANIMED en Allemagne, NISGAT en Italie) et internationaux (ENSAT en Europe). Ces réseaux éditent régulièrement des recommandations et permettent le recueil en grand nombre de tissus biologiques et favorisent l'inclusion des patients dans des essais thérapeutiques.
Cet article propose d'aider l'urologue confronté à un incidentalome surrénalien, en lui proposant, à travers un bilan de malignité, une prise en charge initiale carcinologique. Cela à partir des recommandations établies par le CCAFU en 2018 [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195] et à partir de la littérature scientifique disponible sur PubMed en 2020.
L'exploration d'un IS peut révéler des lésions de nature diverse, affectant bien davantage la corticosurrénale que la médullaire. Les tumeurs malignes de la surrénale (TMS) peuvent être : primitives, touchant le cortex de la glande (carcinome corticosurrénalien [CCS] ou corticosurrénalome malin), ou sa médullaire (phéochromocytome malin) ; ou secondaires à un cancer d'une autre origine (les lymphomes primitifs exceptionnels, majoritairement bilatéraux, ne seront pas traités ici).
|
|
|
Incidentalome surrénalien
|
L'incidence varie de 3 % à plus de 10% chez l'adulte et augmente avec l'âge [2Mayo-Smith W.W., Song J.H., Boland G.L., Francis I.R., Israel G.M., Mazzaglia P.J., et al. Management of Incidental Adrenal Masses: A White Paper of the ACR Incidental Findings Committee. J Am Coll Radiol 2017 ; 14 (8) : 1038-1044 [cross-ref], 3Bhargava P., Sangster G., Haque K., Garrett J., Donato M., D'Agostino H. AMultimodality Review of Adrenal Tumors. Curr Probl Diagn Radiol 2019 ; 48 (6) : 605-615 [cross-ref]]. En cas d'antécédent oncologique, les IS sont plus fréquents (9 à 13 %) [4Blake M.A., Cronin C.G., Boland G.W. Adrenal imaging. AJR Am J Roentgenol 2010 ; 194 (6) : 1450-1460 [cross-ref]]. Les adénomes corticosurrénaliens (75 %) et les myélolipomes (6 %) sont bénins et sont les tumeurs les plus fréquentes. La probabilité qu'un IS soit une tumeur maligne est faible (< 5 %) [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
|
|
|
Carcinome corticosurrénalien (CCS)
|
Cette tumeur est rare, dérivée de la corticosurrénale. Son incidence annuelle est de 0,5 à 2 par million d'habitants. Le CCS est plus fréquent chez la femme (55-60 %) [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. Il peut intervenir à tout âge mais il existe un pic d'incidence entre 40 et 60 ans [5Fassnacht M., Dekkers O.M., Else T., Baudin E., Berruti A., de Krijger R., et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 (4) : G1-G46]. Les CCS sont le plus souvent sporadiques et sont rarement associés à d'autres néoplasies endocriniennes (néoplasie endocrinienne multiple [NEM] de type 1, syndrome de Beckwith-Widman, syndrome de Li-Fraumeni).
Le phéochromocytome (PC) est une tumeur rare neuroendocrine, dérivée des cellules chromaffines de la médullosurrénale. Son incidence annuelle est de 2 à 8 par million d'adultes. Un pic de fréquence est observé entre 30 et 40 ans. Environ 10% des phéochromocytomes sont malins [6Harari A. Inabnet 3rd WB. Malignant pheochromocytoma: a review. Am J Surg 2011 ; 201 (5) : 700-708 [inter-ref]]. La taille supérieure à 5 cm, un envahissement local, une nécrose interne, un pléomorphisme et un hyperchromisme nucléaire sont des caractéristiques suspectes de malignité, mais le seul critère de malignité retenu est l'existence de localisations secondaires dans des organes dépourvus de tissu chromaffine (par ordre de fréquence : les ganglions lymphatiques, les os, le foie, les poumons et les reins) [7Andrade M.O., Cunha V.SD., Oliveira D.C., Moraes O.L., Lofrano-Porto A. What determines mortality in malignant pheochromocytoma? - Report of a case with eighteen-year survival and review of the literature. Arch Endocrinol Metab 2018 ; 62 (2) : 264-269 [cross-ref]].
La majorité des PC malins (PCM) est sporadique (75 %), mais ils peuvent également survenir dans le cadre de maladies génétiques : NEM de type 2 (10 % des PCM sont associés à un syndrome de néoplasie endocrinienne multiple [6Harari A. Inabnet 3rd WB. Malignant pheochromocytoma: a review. Am J Surg 2011 ; 201 (5) : 700-708 [inter-ref]]), maladie de von Hippel-Lindau, mutation de la sous-unité B du succinate déshydrogénase (SDHB), neurofibromatose de type 1, syndrome de Sturge-Weber, sclérose tubéreuse [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195], ce qui justifie qu'en cas de PCM il faille systématiquement écarter un contexte héréditaire. Une consultation oncogénétique est recommandée en cas de suspicion de maladie génétique ou en cas de PC bilatéraux, et chez les patients jeunes (moins de 45 ans) [8Renard J., Clerici T., Licker M., Triponez F. Pheochromocytoma and abdominal paraganglioma. J Visc Surg 2011 ; 148 (6) : e409-16].
|
|
|
Métastase surrénalienne (MS)
|
Chez un patient ayant un antécédent oncologique, un IS est une métastase de son précédent cancer dans 75 % des cas [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]]. Sans antécédent, le risque de métastase est estimé à moins de 2 % en l'absence de tout contexte de cancer actif. S'il existe un contexte de cancer et une atteinte surrénalienne isolée, le risque de métastase est de l'ordre de 30 à 50 %. À l'inverse, si le patient n'a pas de cancer extrasurrénalien connu, l'imagerie est exceptionnellement révélatrice d'un tel primitif [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]].
Tableau de recommandation 1Incidentalome surrénalienRecom mandationsGradeLes IS sont fréquents mais rarement malins. Cependant pour tout IS, un bilan hormonal est recommandé puis un bilan étiologique puis de malignité.FaibleEn cas d'antécédent de cancer extra-surrénalien, l'IS est une métastase jusqu'à preuve du contraire.FaibleLa rareté des tumeurs malignes de la surrénale justifie une prise en charge en centre expert (et en contact avec l'ENS@T) et l'inclusion dès que possible dans des essais thérapeutiques.Faible
| Tableau de recommandation 1 - Incidentalome surrénalien |
|
| Recom mandations |
Grade |
| Les IS sont fréquents mais rarement malins. Cependant pour tout IS, un bilan hormonal est recommandé puis un bilan étiologique puis de malignité. |
Faible |
| En cas d'antécédent de cancer extra-surrénalien, l'IS est une métastase jusqu'à preuve du contraire. |
Faible |
| La rareté des tumeurs malignes de la surrénale justifie une prise en charge en centre expert (et en contact avec l'ENS@T) et l'inclusion dès que possible dans des essais thérapeutiques. |
Faible |
|
Le bilan clinique de l'IS doit être clinique puis hormonal, afin de caractériser son profil de sécrétion (sécrétant ou non). Le bilan étiologique se poursuit en reprenant ou en complétant l'imagerie. En cas d'incidentalome non sécrétant, la crainte est de laisser évoluer, en l'absence de chirurgie, une tumeur maligne asymptomatique. Cette éventualité est faible. Rechercher les arguments de malignité représentera la troisième étape [10Berland L.L., Silverman S.G., Gore R.M., Mayo-Smith W.W., Megibow A.J., Yee J., et al. Managing incidental findings on abdominal CT: white paper of the ACR incidental findings committee. J Am Coll Radiol 2010 ; 7 (10) : 754-773 [cross-ref]].
Il faut rechercher un hypercortisolisme infraclinique (« pré-Cushing ») ou un phéochromocytome passés inaperçus. Les autres sécrétions anormales (tumeur virilisante ou féminisante) sont rarement asymptomatiques et accompagnent exceptionnellement un incidentalome surrénalien [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]].
|
| Interrogatoire et examen clinique |
L'interrogatoire doit rechercher des signes d'hypersécrétion hormonale infraclinique (antécédents, Symptomatologie anodine) (Tableau 1). Il peut aussi relever les conséquences potentielles d'un syndrome de masse abdominale ou des signes évocateurs de malignité (surtout quand ils sont récents et d'installation rapide) : lombalgies profondes, fièvre occulte, anorexie +/- amaigrissement.
L'examen clinique recherche une hypertension artérielle (HTA) et des signes spécifiques des TMS (recherche d'un contact lombaire).
Le tableau clinique des PC (dû à l'excès de catécholamines) peut comprendre : hypertension artérielle (HTA) avec hypokaliémie, perte de poids, pâleur par vasoconstriction périphérique. La triade symptomatique typique dite « triade de Ménard » (céphalées pulsatiles, palpitations cardiaques et tachycardie, sueurs profuses) est inconstante.
Aucun de ces signes cliniques n'oriente vers la malignité du PC [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. La persistance de signes cliniques après la surrénalectomie pour PC est évocatrice de métastases résiduelles et donc de PCM[1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Les PCM sont sécrétants (catécholamines) dans 85 % des cas. Dans ce cas, trois types de complications symptomatiques peuvent survenir : troubles cardiovasculaires (HTA, cardiomyopathie dilatéeâ¦), troubles gastro-intestinaux (constipation extrême) et événements osseux (70 % des patients développent des métastases osseuses, majoritairement lytiques, qui entraînent dans 80 % des cas des douleurs, des fractures ou des compressions médullaires) [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22].
|
| Carcinome corticosurrénalien |
Les CCS, parfois asymptomatiques, sont sécrétants dans 50 à 60 % des cas : Cortisol avec syndrome de Cushing (â 30 %), androgènes avec signes de virilisation chez les femmes (â 20 %), Åstrogènes avec signes de féminisation chez les hommes (â 10 %), sécrétion mixte (â 35 %).
Un tiers des tumeurs virilisantes est malin. Les tumeurs féminisantes sont presque toujours malignes, ce qui représente 10 % des CCS [12Song J.H., Chaudhry F.S., Mayo-Smith W.W. The incidental adrenal mass on CT: prevalence of adrenal disease in 1,049 consecutive adrenal masses in patients with no known malignancy. AJR Am J Roentgenol 2008 ; 190 (5) : 1163-1168 [cross-ref]].
En cas de MS synchrone, les signes cliniques sont dominés par ceux du cancer primitif et d'éventuelles autres localisations. Si la MS est métachrone et isolée, elle est souvent asymptomatique et découverte au cours de la surveillance du cancer primitif [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Avant de réévaluer les critères d'imagerie, il faut réaliser un bilan biologique minimal.
Outre une glycémie à jeun (diabète possible dans les hypercortisolismes et les PC), des dosages hormonaux (Tableau 2) sont recommandés au décours d'une consultation d'endocrinologie (interrogatoire orienté, âge, comorbidités).
Des orientations diagnostiques peuvent être établies par le profil hormonal.
La sécrétion d'hormones sexuelles ou mixtes (Cortisol et hormones sexuelles) est un argument de malignité pour une tumeur de la corticosurrénale.
La méthoxytyramine plasmatique est un marqueur prédictif de malignité des PC [13Berruti A., Baudin E., Gelderblom H., Haak H.R., Porpiglia F., Fassnacht M., et al. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up Ann Oncol 2012 ; 23 : vii131-8, 14Eisenhofer G., Lenders J.W., Siegert G., Bornstein S.R., Friberg P., Milosevic D., et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status Eur J Cancer 2012 ; 48 (11) : 1739-1749 [cross-ref]]. Un outil pourrait être utile pour différencier une tumeur surrénalienne corticale bénigne d'une maligne : le profil stéroïdien urinaire mesuré par spectrométrie de masse (GC-MS ou LC-MS) [15Kerkhofs T.M., Kerstens M.N., Kema I.P., Willems T.P., Haak H.R. Diagnostic Value of Urinary Steroid Profiling in the Evaluation of Adrenal Tumors. Horm Cancer 2015 ; 6 (4) : 168-175 [cross-ref]].
|
| Bilan biologique complémentaire |
Un bilan génétique n'est réalisé qu'en cas d'orientation vers un contexte héréditaire (antécédents familiaux, patients jeunes, tumeurs bilatérales, localisation extra-surrénalienne, PCM, CSS).
La taille d'un IS peut être prédictive de sa malignité quelle que soit la modalité d'imagerie. Cependant, une étude récente semble réduire la valeur prédictive de CCS du seul critère de taille [17Kahramangil B, Kose E, Remer EM, Reynolds JP, Stein R, Rini B, et al. A Modern Assessment of Cancer Risk in Adrenal Incidentalomas: Analysis of 2219 Patients. Ann Surg 2020.].
Au-delà de 6 cm, la proportion de tumeurs malignes est de 25 %, alors qu'elle est inférieure à 2 % pour les masses de moins de 4 cm [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref],18Vural V., Kilinc E.M., Saridemir D., Gok I.B., Huseynov A., Akbarov A., et al. Association between Tumor Size and Malignancy Risk in Hormonally Inactive Adrenal Incidentalomas. Cureus 2020 ; 12 (1) : e6574]. Un diamètre tumoral > 6 cm est donc dans tous les cas un argument de malignité.
Compte tenu de la croissance tumorale généralement rapide des TMS, il a été proposé pour les incidentalomes surveillés de contrôler la tomodensitométrie (TDM) à 6 mois et 1 an. En cas de stabilité et d'IS non sécrétant, il n'y a pas d'argument pour recommander un suivi radiologique systématique [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195,19Sebe P., Rigaud J., Avances C., Brunaud L., Caillard C., Camparo P., et al. [CCAFU's contribution to the French National Cancer Institute's reference frame: Adrenal malignant tumors] Prog Urol 2013 ; 23 : S167-74], mais il faut surveiller l'apparition de symptômes ou de signes cliniques.
Pour les CCS, la spécificité est respectivement de 52, 80, 95 et 98 % pour des diamètres > 4 cm, > 6 cm, > 8 cm et > 10 cm [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Pour les PCM, la spécificité est respectivement de 20, 65 et 89 % pour des diamètres 4 cm, 6 cm et 8 cm [20Shen W.T., Sturgeon C., Clark O.H., Duh Q.Y., Kebebew E. Should pheochromocytoma size influence surgical approach? A comparison of 90 malignant and 60 benign pheochromocytomas Surgery 2004 ; 136 (6) : 1129-1137 [cross-ref]].
|
| Caractéristiques de la tomodensitométrie (TDM) |
L'étude de la densité spontanée différencie les adénomes, riches en graisse microscopique, des lésions malignes, plus pauvres, avec une sensibilité et une spécificité de 71 et 98 %, respectivement [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]]. Ainsi, les tumeurs malignes auraient une densité spontanée plus importante que les tumeurs bénignes. Dans la série de Szolar et al., les densités moyennes des tumeurs surrénaliennes sont : 39 UH pour les CCS, 44 UH pour les PCM et 34 UH pour les métastases surrénaliennes (contre 8 UH pour les adénomes surrénaliens). Cette tendance serait confirmée sur les densités 10 minutes après injection [21Szolar D.H., Korobkin M., Reittner P., Berghold A., Bauernhofer T., Trummer H., et al. Adrenocortical carcinomas and adrenal pheochromocytomas: mass and enhancement loss evaluation at delayed contrast-enhanced CT. Radiology 2005 ; 234 (2) : 479-485 [cross-ref]].
L'analyse du rehaussement de la masse, après injection, aide également à la caractérisation. Les adénomes présentent un wash-out absolu (tenant compte de la densité spontanée) ou relatif plus important que celui des métastases (hors métastases hypervasculaires de carcinome rénal à cellules claires). Le calcul du wash-out ne permet pas de différencier l'adénome du phéochromocytome, tumeur hypervasculaire.
Kahramangil confirme que la suspicion de malignité des IS résulte d'un faisceau d'arguments issu des évaluations clinique, hormonale et par imagerie [17Kahramangil B, Kose E, Remer EM, Reynolds JP, Stein R, Rini B, et al. A Modern Assessment of Cancer Risk in Adrenal Incidentalomas: Analysis of 2219 Patients. Ann Surg 2020.].
|
| Caractéristiques de l'imagerie par résonance magnétique (IRM) |
L'IRM abdominale n'apporte pas d'éléments diagnostiques supplémentaires par rapport au scanner (sa sensibilité et sa spécificité sont moindres : 78 et 87 %, respectivement dans la caractérisation tissulaire [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]]). La prise de contraste après injection de gadolinium est importante avec un hypersignal évocateur mais non spécifique des PCM sur les séquences T2. La grande lenteur du wash-out, après injection de gadolinium, est évocatrice des CCS. Les lésions bénignes tissulaires ont, en T1 et T2, une intensité de signal égale ou légèrement inférieure à celle du foie normal [23Wu D., Tischler A.S., Lloyd R.V., DeLellis R.A., de Krijger R., van Nederveen F., et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol 2009 ; 33 (4) : 599-608 [cross-ref]]. Le phéochromocytome présente un hypersignal T2, des remaniements hémorragiques peuvent modifier le signal : rapport surrénale/foie > 3 et un rehaussement rapide et intense à l'injection de gadolinium.
En séquences classiques, l'IRM apprécierait mieux l'envahissement local et veineux d'une tumeur potentiellement maligne, grâce à la meilleure résolution en contraste, mais la résolution spatiale du scanner est meilleure. Elle peut donc théoriquement compléter les données du scanner abdominal pour affiner le bilan d'extension locorégional, métastatique, vasculaire ou ganglionnaire. Même dans ces indications, l'amélioration actuelle des TDM limite la place de l'IRM dans de nombreuses indications. Elle est par contre un outil de surveillance chez les patients jeunes, pour limiter une irradiation itérative.
La séquence phase-opposition de phase ou spectroscopie par résonance magnétique est utile. Une diminution du signal en opposition de phase (eau + graisse / graisse - eau) de 20 % serait en faveur d'un adénome [24Gaujoux S., Mihai R. joint working group of E, Ensat. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma Br J Surg 2017 ; 104 (4) : 358-376 [cross-ref]], le plus souvent riche en graisse microscopique (spécificité et sensibilité élevées [25Platzek I., Sieron D., Plodeck V., Borkowetz A., Laniado M., Hoffmann R.T. Chemical shift imaging for evaluation of adrenal masses: a systematic review and meta-analysis. Eur Radiol 2019 ; 29 (2) : 806-817 [cross-ref]]).
Tableau de recommandation 2Bilan radiologique des tumeurs surrénaliennesRecommandationsGradeLe risque de malignité d'une TS s'accroît avec la taille de la lésion.FortLors de la surveillance d'un IS : une croissance rapide d'une TS est suspecte de malignité.FortUn IS avec un bilan hormonal normal et dont la TDM montre une densité < 10 UH, avec un parenchyme homogène et un wash-out rapide, est considéré comme un adénome bénin. Quand il est inférieur à 4 cm, aucun suivi paraclinique n'est nécessaire [16].FortUn IS non sécrétant > 4 cm, sans critère péjoratif au scanner non injecté peut justifier d'un bilan d'imagerie complémentaire, être surveillé par scanner ou IRM dans l'année, ou être opéré d'emblée [16].FortEn TDM : une densité spontanée > 20 UH, un aspect hétérogène, une hypervascularisation, un wash-out réduit et/ou des limites irrégulières (envahissement local) sont des arguments à risque de malignité pour une TS.FortDans les TS, l'IRM est un examen de deuxième intention :•IS avec contre-indication à la TDM injectée.•TS atypique au scanner (densité > 10 UH) : une chute de signal en opposition de phase permet d'affirmer le caractère bénin de la tumeur [26].•TS suspect de malignité (pour préciser l'envahissement local et à distance).FortLégende : TS = tumeur surrénalienne, IS = incidentalome malin ; UH = Unités Hounsfield ;
| Tableau de recommandation 2 - Bilan radiologique des tumeurs surrénaliennes |
|
| Recommandations |
Grade |
| Le risque de malignité d'une TS s'accroît avec la taille de la lésion. |
Fort |
| Lors de la surveillance d'un IS : une croissance rapide d'une TS est suspecte de malignité. |
Fort |
| Un IS avec un bilan hormonal normal et dont la TDM montre une densité < 10 UH, avec un parenchyme homogène et un wash-out rapide, est considéré comme un adénome bénin. Quand il est inférieur à 4 cm, aucun suivi paraclinique n'est nécessaire [16Fassnacht M., Arlt W., Bancos I., Dralle H., Newell-Price J., Sahdev A., et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016 ; 175 (2) : G1-G34]. |
Fort |
| Un IS non sécrétant > 4 cm, sans critère péjoratif au scanner non injecté peut justifier d'un bilan d'imagerie complémentaire, être surveillé par scanner ou IRM dans l'année, ou être opéré d'emblée [16Fassnacht M., Arlt W., Bancos I., Dralle H., Newell-Price J., Sahdev A., et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016 ; 175 (2) : G1-G34]. |
Fort |
| En TDM : une densité spontanée > 20 UH, un aspect hétérogène, une hypervascularisation, un wash-out réduit et/ou des limites irrégulières (envahissement local) sont des arguments à risque de malignité pour une TS. |
Fort |
Dans les TS, l'IRM est un examen de deuxième intention :
|
• |
IS avec contre-indication à la TDM injectée. |
|
• |
TS atypique au scanner (densité > 10 UH) : une chute de signal en opposition de phase permet d'affirmer le caractère bénin de la tumeur [26Libe R., Assie G. [Adrenocortical carcinoma: Update in 2014] Presse Med 2014 ; 43 : 401-409 [cross-ref]].
|
|
• |
TS suspect de malignité (pour préciser l'envahissement local et à distance). |
|
Fort |
|
| Légende : TS = tumeur surrénalienne, IS = incidentalome malin ; UH = Unités Hounsfield ; |
|
Kahramangil confirme que la suspicion de malignité des IS est un faisceau d'arguments issu de l'évaluation clinique, des explorations hormonales et des caractéristiques d'imagerie [17Kahramangil B, Kose E, Remer EM, Reynolds JP, Stein R, Rini B, et al. A Modern Assessment of Cancer Risk in Adrenal Incidentalomas: Analysis of 2219 Patients. Ann Surg 2020.]. Certaines hypothèses diagnostiques peuvent aussi être évoquées, qui pourront justifier une exploration scintigraphique (fonctionnelle) pour compléter le bilan et/ou confirmer le diagnostic.
|
| Caractéristiques scintigraphiques |
|
| Tomographie par émission de positons (TEP) au fluorodésoxyglucose (18F) ou 2-désoxy-2-(18F) fluoro-D-glucose (18F-FDG) |
Devant une lésion surrénalienne hypermétabolique au 18F-FDG, il faut évoquer quatre diagnostics principaux : un PC, un CCS, une MS ou un lymphome.
En cas de suspicion de CCS, la TEP-18FDG est l'examen scintigraphique de référence, tant à la phase diagnostique que pour le suivi [24Gaujoux S., Mihai R. joint working group of E, Ensat. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma Br J Surg 2017 ; 104 (4) : 358-376 [cross-ref],27Libe R. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Front Cell Dev Biol 2015 ; 3 : 45]. La TEP est également utile dans le bilan d'extension des CCS pour le diagnostic des métastases à distance.
Cet examen permet de calculer le rapport de la SUV max (maximal Standard Uptake Value ) de la tumeur sur celui du foie (SUV max tumeur/SUV max foie). Un rapport ⥠1,45 est fortement prédictif de malignité [28Thompson L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002 ; 26 (5) : 551-566 [cross-ref]].
La TEP au 18F-FDG semble avoir un intérêt différent pour caractériser les tumeurs de la médulla, car il est connu que les PC bénins comme malins peuvent présenter une captation du 18F-FDG [29Tenenbaum F., Lataud M., Groussin L. [Update in adrenal imaging] Presse Med 2014 ; 43 : 410-419 [inter-ref]]. Elle est maintenant recommandée dans le bilan d'extension des phéochromocytomes malins en préopératoire [30Plouin P.F., Amar L., Dekkers O.M., Fassnacht M., Gimenez-Roqueplo A.P., Lenders J.W., et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016 ; 174 (5) : G1-G10]. Elle est maintenant autorisée en France, devant la découverte d'un incidentalome surrénalien [31Taïeb D., Salaün P.Y. Recommandations de bonne pratique clinique pour l'utilisation de la TEP en cancérologie Incidentalomes surrenaliens. Médecine Nucléaire 2019 ; 43 : 66-68].
|
| Tomographie par émission de positons au 18F-DOPA (Fluoro-18-L-Dihydroxyphenylalanine) |
En cas de suspicion de PC, le traceur de choix est le 18F-DOPA. Il est utile au diagnostic positif et à la mise en évidence des éventuelles localisations secondaires, avec une sensibilité proche de 100 %. Il est plus sensible que le MIBG et peut être couplé au 18-FDG [32Taïeb D., Timmers H.J., Hindie E., Guillet B.A., Neumann H.P., Walz M.K., et al. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging 2012 ; 39 (12) : 1977-1995].
|
| Scintigraphie au Méta-iodo-benzylguanidine marquée à l'iode 123 (123I-MIBC) |
Traceur spécifique de la médullosurrénale, la fixation du 123I-MIBG, en regard d'une masse surrénalienne, est très en faveur d'un PC [29Tenenbaum F., Lataud M., Groussin L. [Update in adrenal imaging] Presse Med 2014 ; 43 : 410-419 [inter-ref]]. Classiquement utilisée depuis les années 1980 pour confirmer le diagnostic de PC, elle peut être utile quand les dérivés méthoxylés sont limites ou variables d'un prélèvement à un autre. Quand le diagnostic de PC est posé, elle élimine aussi les autres localisations ou de rares métastases [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]] mais, dans ce cas, elle est progressivement supplantée par la TEP (elle garde seulement une indication quand la TEP est indisponible).
En cas de PCM, son utilisation ne devrait se limiter qu'aux malades pouvant répondre à une radiothérapie interne (ou radiothérapie métabolique) à l'iode 131 (131l), vectorisée au MIBG (prédictive de l'efficacité de cette radiothérapie métabolique) [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22,33Lenders J.W., Duh Q.Y., Eisenhofer G., Gimenez-Roqueplo A.P., Grebe S.K., Murad M.H., et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014 ; 99 (6) : 1915-1942 [cross-ref]].
Tableau de recommandation 3Imagerie fonctionnelle d'un incidentalome surrénalienBilan d'un IS par imagerie fonctionnelleGradeLa TEP au 18F-FDG est recommandée systématiquement en cas de suspicion de CSS et d'IS avec antécédent de cancer extra-surrénalien.ModéréQuand un PCM est suspecté, une TEP au 18F-FDG est recommandée et peut être couplée au 18F-DOPA.FaibleDans les PCM, l'utilisation de la scintigraphie au 123I-MIBG se limite aux candidats à une radiothérapie métabolique (131l vectorisé par ce marqueur).Modéré
| Tableau de recommandation 3 - Imagerie fonctionnelle d'un incidentalome surrénalien |
|
| Bilan d'un IS par imagerie fonctionnelle |
Grade |
| La TEP au 18F-FDG est recommandée systématiquement en cas de suspicion de CSS et d'IS avec antécédent de cancer extra-surrénalien. |
Modéré |
| Quand un PCM est suspecté, une TEP au 18F-FDG est recommandée et peut être couplée au 18F-DOPA. |
Faible |
| Dans les PCM, l'utilisation de la scintigraphie au 123I-MIBG se limite aux candidats à une radiothérapie métabolique (131l vectorisé par ce marqueur). |
Modéré |
|
|
|
|
Place de la biopsie percutanée
|
La place de la biopsie percutanée est très limitée. Elle n'est pas recommandée chez un patient sans antécédent néoplasique [34Tabarin A., Bardet S., Bertherat J., Dupas B., Chabre O., Hamoir E., et al. Exploration and management of adrenal incidentalomas. French Society of Endocrinology Consensus. Ann Endocrinol (Paris) 2008 ; 69 (6) : 487-500 [inter-ref]]. La seule indication est une suspicion de métastase surrénalienne ou celle d'un lymphome ou d'un sarcome rétropéritonéal [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Même dans ces circonstances, il faut d'abord écarter formellement un PC infraclinique car la prévalence de ce dernier chez un patient porteur d'un cancer extra-surrénalien est relativement élevée, de 5 à 25 % [9Menegaux F., Chereau N., Peix J.L., Christou N., Lifante J.C., Paladino N.C., et al. Management of adrenal incidentaloma. J Visc Surg 2014 ; 151 (5) : 355-364 [inter-ref]]. Elle peut être exceptionnellement nécessaire (après avoir écarté un PC) pour confirmer un diagnostic d'une lésion surrénalienne d'emblée métastatique irrésécable (elle doit alors être associée à un marquage immunohistochimie anti-SF1). En cas de suspicion de PCM, elle est classiquement contreindiquée (risque d'hypertension maligne sur décharge de catécholamines) [16Fassnacht M., Arlt W., Bancos I., Dralle H., Newell-Price J., Sahdev A., et al. Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2016 ; 175 (2) : G1-G34].
En cas de suspicion de CCS, elle est contre-indiquée [24Gaujoux S., Mihai R. joint working group of E, Ensat. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma Br J Surg 2017 ; 104 (4) : 358-376 [cross-ref]], du fait du risque de dissémination tumorale, liée à la rupture capsulaire [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. Cependant, elle peut avoir un intérêt en cas de doute entre une métastase surrénalienne et un CCS dès lors que l'on est sûr de son caractère non sécrétant [35Fassnacht M., Libe R., Kroiss M., Allolio B. Adrenocortical carcinoma: a clinician's update. Nat Rev Endocrinol 2011 ; 7 (6) : 323-335 [cross-ref]].
|
|
|
Prise en charge thérapeutique
|
Les recommandations thérapeutiques sont présentées sous forme d'algorithmes décisionnels, tirés des recommandations du CCAFU éditées en 2018 [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. Le premier propose la prise en charge d'un IS en fonction des résultats du bilan de malignité (Figure 1). Des algorithmes spécifiques sont proposés en cas de suspicion de CCS (Figure 2) et de suspicion de PCM (Figure 3).
Figure 1.
Algorithme décisionnel des IS [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
(Abréviations : TEP 18FDG = tomodensitométrie par émission de positons au fluorodésoxyglucose (18F) ; CCS = carcinome corticosurrénalien ; PCM = phéochromocytome malin).
Figure 2.
Algorithme décisionnel des CCS [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Abréviations : ttt = traitement ; R0 = pas de tumeur résiduelle ; R1 = reliquat tumoral microscopique.
Figure 3.
Algorithme décisionnel des PCM [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Abréviations : PCM = phéochromocytome malin ; HTA = hypertension artérielle ; ttt = traitement.
|
|
|
Bilan carcinologique postopératoire
|
|
|
|
Carcinome corticosurrénalien
|
|
| Le score histopronostique de Weiss |
En cas de diagnostic douteux de CCS, les lames doivent être relues par un pathologiste spécialisé, dans le cadre du réseau COMETE. Une double lecture est recommandée au sein du réseau.
En cas de doute sur l'origine corticale ou non de la tumeur, la recherche immunohistochimique de l'expression du steroidogenic factor-1 (SF-1) doit être systématique car il s'agit du marqueur le plus sensible et spécifique de la corticosurrénale [36Sbiera S., Schmull S., Assie G., Voelker H.U., Kraus L., Beyer M., et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J Clin Endocrinol Metab 2010 ; 95 (10) : E161-71].
Dans les corticosurrénalomes avérés, l'examen anatomopathologique est capital pour établir le diagnostic de corticosurrénalome. Il permet l'établissement final du stade pTNM, le statut de résection « R » et surtout le calcul du score histopronostique de Weiss (Tableau 3). Le diagnostic de malignité est donc retenu quand une tumeur localisée présente un score de Weiss ⥠3 et/ou en cas d'envahissement local ou de métastases à distance [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195,5Fassnacht M., Dekkers O.M., Else T., Baudin E., Berruti A., de Krijger R., et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 (4) : G1-G46,26Libe R., Assie G. [Adrenocortical carcinoma: Update in 2014] Presse Med 2014 ; 43 : 401-409 [cross-ref], 27Libe R. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Front Cell Dev Biol 2015 ; 3 : 45,37Baudin E. Endocrine Tumor Board of Gustave R. Adrenocortical carcinoma. Endocrinol Metab Clin North Am 2015 ; 44 (2) : 411-434 [inter-ref]].
|
| Classification TNM des CCS |
La 8e édition de la TNM (2016) ne présente pas de mise à jour par rapport à la version de 2009 (Tableau 4) [38Fassnacht M., Johanssen S., Quinkler M., Bucsky P., Willenberg H.S., Beuschlein F., et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer 2009 ; 115 (2) : 243-250 [cross-ref], 39Amin M.B., Greene F.L., Edge S.B., Compton C.C., Gershenwald J.E., Brookland R.K., et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more âpersonalizedâ approach to cancer staging. CA Cancer J Clin 2017 ; 67 (2) : 93-99 [cross-ref]]. Elle a une valeur pronostique et est utile à la prise en charge thérapeutique.
Le diagnostic histologique de PC ne pose en général pas de problème mais les critères de malignité sont débattus [40Patey M. [Pheochromocytoma and the diagnosis of malignancy: recent data and the role of the pathologist] Ann Pathol 2008 ; 28 (1) : S42-4 [inter-ref]]. l'invasion capsulaire et l'invasion vasculaire sont considérées comme des critères à haut risque de malignité mais ne sont pas toujours associées à une maladie métastatique [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195,22Ng C.S., Altinmakas E., Wei W., Ghosh P., Li X., Grubbs E.G., et al. Utility of Intermediate-Delay Washout CT Images for Differentiation of Malignant and Benign Adrenal Lesions: A Multivariate Analysis. AJR Am J Roentgenol 2018 ; 211 (2) : W109-W115 [inter-ref]].
Actuellement, la seule preuve formelle de malignité est l'envahissement des organes de voisinage ou les métastases à distance [6Harari A. Inabnet 3rd WB. Malignant pheochromocytoma: a review. Am J Surg 2011 ; 201 (5) : 700-708 [inter-ref],23Wu D., Tischler A.S., Lloyd R.V., DeLellis R.A., de Krijger R., van Nederveen F., et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol 2009 ; 33 (4) : 599-608 [cross-ref],40Patey M. [Pheochromocytoma and the diagnosis of malignancy: recent data and the role of the pathologist] Ann Pathol 2008 ; 28 (1) : S42-4 [inter-ref]].
Bien que non validé (et parfois discordant avec d'autres critères immunohistochimiques), le score pronostique le plus utilisé est le Pheochromocytoma of the Adrenal gland Scaled Score (PASS), proposé en 2002 par Thompson [28Thompson L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002 ; 26 (5) : 551-566 [cross-ref]]. Le score repose sur les items recensés dans le Tableau 5. Un score inférieur à 4 est en faveur de la bénignité ; supérieur à 6 de la malignité. Il n'est pas recommandé en pratique courante [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Les cancers primitifs sont en premier lieu : cancer du poumon (35 %), cancer du rein, cancer du sein, mélanome malin, cancer de l'estomac, cancer colorectal et lymphome. Une localisation surrénalienne isolée est rare, mais reste souvent confinée à la glande, d'où l'intérêt d'une chirurgie d'exérèse [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195].
Les métastases surrénaliennes sont classées M+ dans la classification TNM du cancer primitif.
Tableau de recommandation 4Recommandation sur l'histologie des tumeurs surrénaliennesRecommandationsGradeLe diagnostic de certitude du CCS est histologique, permettant d'établir la classification TNM.FortPour les PC : aucun critère histologique n'est spécifique de son caractère malin, en dehors de l'envahissement des organes de voisinage.FortPour les PC : l'invasion capsulaire et l'invasion vasculaire sont des facteurs de risque inconstants de malignité. Le score PASS est imprécis et son utilisation n'est donc pas recommandée en pratique courante.FaibleIl n'y a pas de classification TNM pour les PCM.Fort
| Tableau de recommandation 4 - Recommandation sur l'histologie des tumeurs surrénaliennes |
|
| Recommandations |
Grade |
| Le diagnostic de certitude du CCS est histologique, permettant d'établir la classification TNM. |
Fort |
| Pour les PC : aucun critère histologique n'est spécifique de son caractère malin, en dehors de l'envahissement des organes de voisinage. |
Fort |
| Pour les PC : l'invasion capsulaire et l'invasion vasculaire sont des facteurs de risque inconstants de malignité. Le score PASS est imprécis et son utilisation n'est donc pas recommandée en pratique courante. |
Faible |
| Il n'y a pas de classification TNM pour les PCM. |
Fort |
|
|
|
|
Carcinome corticosurrénalien
|
Un âge avancé et la sécrétion de Cortisol semblent associés à un pronostic plus sévère [26Libe R., Assie G. [Adrenocortical carcinoma: Update in 2014] Presse Med 2014 ; 43 : 401-409 [cross-ref]].
Plusieurs marqueurs moléculaires pourraient aussi être associés à un mauvais pronostic. Ils sont mesurés en immunohistochimie (accumulation nucléaire de p53, forte intensité du marquage de SF-1, accumulation nucléaire de la bêtacaténine), ou par des techniques de biologie moléculaire (profil d'expression des gènes en faveur d'une tumeur de mauvais pronostic, niveau de méthylation élevé de la région promotrice des gènes, combinaison particulière de pertes et de gains chromosomiques) sans que leur utilisation, en pratique courante, soit recommandée [26Libe R., Assie G. [Adrenocortical carcinoma: Update in 2014] Presse Med 2014 ; 43 : 401-409 [cross-ref]].
La proportion de cellule en mitose (grade) est un critère d'agressivité reconnu en histologie. Dans le CCS, le critère pronostic histologique reconnu est le grade exprimé par l'analyse d'un marquage immunohistochimique dirigé contre le Ki-67. Les seuils définissant un pronostic péjoratif sont : > 10 % : haut risque ; > 30 % : très haut risque. Les index de prolifération, l'appréciation de la qualité de la résection, le statut ganglionnaire apportent également des informations sur le pronostic.
|
| Classifications pronostiques |
La classification European Network for the Study of Adrenal Tumors (ENS@T) [38Fassnacht M., Johanssen S., Quinkler M., Bucsky P., Willenberg H.S., Beuschlein F., et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer 2009 ; 115 (2) : 243-250 [cross-ref]] est superposable à celle de l'AJCC, regroupant les CCS en quatre groupes pronostiques par agrégation de la TNM (Tableau 6) [39Amin M.B., Greene F.L., Edge S.B., Compton C.C., Gershenwald J.E., Brookland R.K., et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more âpersonalizedâ approach to cancer staging. CA Cancer J Clin 2017 ; 67 (2) : 93-99 [cross-ref]].
La survie à 5 ans est différente selon ces stades : 66-82 % pour les stades 1, 58-64 % pour les stades 2, 24-50 % pour les stades 3 et 0-17 % pour les stades 4 [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195,13Berruti A., Baudin E., Gelderblom H., Haak H.R., Porpiglia F., Fassnacht M., et al. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up Ann Oncol 2012 ; 23 : vii131-8].
La résection en marges saines est une condition indispensable à la survie à long terme [1Savoie P.H., Murez T., Flechon A., Sebe P., Rocher L., Camparo P., et al. French ccAFU guidelines - Update 2018-2020: Adrenal cancer Prog Urol 2018 ; 28 : R177-R195]. De même, la rupture capsulaire est prédictive de récidive.
Un groupe d'experts américains (United States ACC Study Group) a proposé de modifier la classification TNM en y incluant l'invasion lymphovasculaire, ce qui améliorerait la valeur pronostique principalement dans les stades T2 et T3 [41Crona J., Beuschlein F., Pacak K., Skogseid B. Advances in adrenal tumors 2018. Endocr Relat Cancer 2018 ; 25 (7) : R405-R420, 42Poorman C.E., Ethun C.G., Postlewait L.M., Tran T.B., Prescott J.D., Pawlik T.M., et al. A Novel T-Stage Classification System for Adrenocortical Carcinoma: Proposal from the US Adrenocortical Carcinoma Study Group. Ann Surg Oncol 2018 ; 25 (2) : 520-527 [cross-ref]].
Aucun facteur pronostique validé n'existe pour le PCM. La survie à 5 ans des PCM s'étend de 40 à 77 % [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22].
La croissance tumorale est le motif principal de décès par PCM. Le contrôle tumoral doit donc être l'objectif principal de la prise en charge des PCM. Cependant, les manifestations cliniques, dues à l'excès de catécholamines (hypertension artérielle, constipationâ¦), doivent être traitées car elles seraient responsables de 30 % des décès par PCM [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22]. Enfin, le décès consécutif à un autre cancer, notamment dans un contexte de maladie génétique (NEM) est possible [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22,43Ayala-Ramirez M., Feng L., Habra M.A., Rich T., Dickson P.V., Perrier N., et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer 2012 ; 118 (11) : 2804-2812 [cross-ref]].
Pour le PCM, plusieurs facteurs péjoratifs ont cependant été proposés : un volume tumoral important, l'existence ou le nombre de métastases viscérales et la présence d'une mutation du gène succinate déshydrogénase B (SDHB) [11Baudin E., Habra M.A., Deschamps F., Cote G., Dumont F., Cabanillas M., et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol 2014 ; 171 (3) : R111-22].
Tableau de recommandation 5Recommandations pronostiques des tumeurs surrénaliennesRecommandationsGradeLes CCS de haut grade (> 20 mitoses/50ch) et/ou l'expression immunohistochimique du Ki-67 sont des arguments de malignité associés à un pronostic péjoratif.FaibleLe compte-rendu anatomopathologique d'un CSS doit préciser au minimum : le score de Weiss, le stade T, le statut N, la qualité des marges (R) et l'expression exacte en % du Ki-67.FortPour le CCS, la classification ENS@T (tirée de la TNM) est recommandée et a un intérêt majeur car elle a une valeur pronostique et oriente le traitement.FortDans les PCM, il n'existe aucun facteur pronostique validé. Cependant certains facteurs sont péjoratifs (volume tumoral important, métastases viscérales, mutation SDHB).Faible
| Tableau de recommandation 5 - Recommandations pronostiques des tumeurs surrénaliennes |
|
| Recommandations |
Grade |
| Les CCS de haut grade (> 20 mitoses/50ch) et/ou l'expression immunohistochimique du Ki-67 sont des arguments de malignité associés à un pronostic péjoratif. |
Faible |
| Le compte-rendu anatomopathologique d'un CSS doit préciser au minimum : le score de Weiss, le stade T, le statut N, la qualité des marges (R) et l'expression exacte en % du Ki-67. |
Fort |
| Pour le CCS, la classification ENS@T (tirée de la TNM) est recommandée et a un intérêt majeur car elle a une valeur pronostique et oriente le traitement. |
Fort |
| Dans les PCM, il n'existe aucun facteur pronostique validé. Cependant certains facteurs sont péjoratifs (volume tumoral important, métastases viscérales, mutation SDHB). |
Faible |
|
|
|
|
Carcinome corticosurrénalien
|
Après résection complète, une surveillance clinique, hormonale et par imagerie (scanner thoraco-abdomino-pelvien ou 18FDG-TEP) tous les 3 mois pendant 2 ans, puis tous les 3 à 6 mois pendant 3 ans est recommandée [5Fassnacht M., Dekkers O.M., Else T., Baudin E., Berruti A., de Krijger R., et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 (4) : G1-G46]. Au-delà de 5 ans, la surveillance est raisonnable mais doit être envisagée au cas par cas.
Pour les CCS avancés, le protocole de surveillance dépend des facteurs pronostiques, de l'efficacité attendue du traitement et de la toxicité liée au traitement, ainsi que des options thérapeutiques alternatives disponibles.
Chez tous les patients, un bilan hormonal régulier est recommandé [5Fassnacht M., Dekkers O.M., Else T., Baudin E., Berruti A., de Krijger R., et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 (4) : G1-G46].
Le suivi des PCM repose sur des dosages plasmatiques ou urinaires de métanéphrines [33Lenders J.W., Duh Q.Y., Eisenhofer G., Gimenez-Roqueplo A.P., Grebe S.K., Murad M.H., et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014 ; 99 (6) : 1915-1942 [cross-ref]]. Un rythme semestriel la première année puis au minimum annuel pendant 5 ans est conseillé. La récidive peut survenir très tardivement et un terrain génétique peut rester non diagnostiqué, suggérant la nécessité d'un suivi à vie [8Renard J., Clerici T., Licker M., Triponez F. Pheochromocytoma and abdominal paraganglioma. J Visc Surg 2011 ; 148 (6) : e409-16]. Il est possible d'espacer les surveillances avec le temps (tous les 6 mois au-delà de 10 ans) [6Harari A. Inabnet 3rd WB. Malignant pheochromocytoma: a review. Am J Surg 2011 ; 201 (5) : 700-708 [inter-ref]]. Une TEP-18 FDG +/- DOPA est recommandée au moindre doute.
L'éradication d'une MS n'influence pas le suivi spécifique du cancer primitif. En cas de traitement conservateur, une imagerie par TEP-18 FDG peut compléter le suivi habituel.
Les IS sont fréquents mais rarement malins. La prise en charge des TMS, hautement spécifique, justifie donc en France leur référencement au réseau COMETE (Annexe 1). Un bilan minimal endocrinien est nécessaire même en cas de suspicion d'atteinte secondaire. La prise en charge diffère selon l'orientation diagnostique. Comme pour toute pathologie rare, la prise en charge dans un centre en ayant l'expertise est recommandée.
|
|
|
Déclaration de liens d'intérêts
|
P.-H. Savoie : Orateur et consultant pour le CCAFU online (AFU), Consultant pour BMS (membre d'un sous-comité), Janssen (expert urologue), Co-investigateur : Novartis, Janssen-Cilag, Intervention aux JOUM (CCAFU).
Les autres auteurs déclarent ne pas avoir de liens d'intérêts.
|
|
|
Annexe 1. Liste des centres de référence COMETE - cancers de la surrénale
|
|
• |
Centre expert national de référence (cocoordination) : Paris - Villejuif et centres associés (région Île-de-France) |
Responsables : Pr Bertherat (Cochin), Dr Baudin (institut Gustave-Roussy)
Centres associés en Île-de-France (contacts) : HEGP (Dr Amar), Saint-Antoine (Dr Donadille), Ambroise-Paré (Pr Raffin-Sanson), Pitié-Salpetrière (Dr Ghander), Saint-Louis (Dr Chougnet), Reims (Pr Delemer)
|
• |
Neuf centres experts régionaux - (centres associés) |
|
â |
Centre de compétences des cancers de la surrénale - région des Pays de la Loire : Angers |
Responsable : Pr Rohmer, CHU d'Angers
Centres associés (contacts) : Brest (Pr Kerlan), Tours (Dr Pierre), Nantes (Dr Drui)
|
â |
Centre de compétences des cancers de la surrénale - région Bourgogne : Dijon |
Responsables : Pr Vergès et Dr Zanetta, CHU Dijon Bourgogne - hôpital François-Mitterrand
Centre associé (contact) : Besançon (Dr Schillo)
|
â |
Centre de compétences des cancers de la surrénale - région Rhône-Alpes : Grenoble |
Responsable : Pr Chabre, CHU de Grenoble site Nord - hôpital Albert-Michallon
Centres associés (contacts) : Clermont-Ferrand (Pr Tauveron), Lyon (Pr Borson-Chazot)
|
â |
Centre de compétences des cancers de la surrénale - région Nord-Pas-de-calais : Lille |
Responsable : Pr Vantyghem, CHRU de Lille - hôpital Claude-Huriez
Centre associé (contact) : Amiens (Pr Desailloud)
|
â |
Centre de compétences des cancers de la surrénale - région PACA : Marseille |
Responsable : Pr Niccoli, CHU de Marseille - hôpital de la Timone
Centres associés (contacts) : Marseille (Pr Castinetti), Montpellier (Dr Raingeard), Nice (Pr Sadoul)
|
â |
Centre de compétences des cancers de la surrénale - région Aquitaine : Bordeaux |
Responsable : Pr Tabarin, CHU de Bordeaux-GH Sud - hôpital Haut-Lévêque
Centres associés (contacts) : Limoges (Pr Archambeaud), Poitiers (PrMarechaud), Pointe-à-Pitre (Dr Cephise), Papeete (Dr Rachedi), La Réunion (Dr Lemoullec)
|
â |
Centre de compétences des cancers de la surrénale, région Haute-Normandie : Rouen |
Responsable : Pr Lefebvre, CHU de Rouen
Centre associé (contact) : Caen (Pr Reznik)
|
â |
Centre de compétences des cancers de la surrénale - région Alsace : Strasbourg |
Responsable : Pr Goichot, CHU de Strasbourg - hôpital de Hautepierre
Centre associé (contact) : Nancy (Pr Brunaud)
|
â |
Centre de compétences des cancers de la surrénale - région Midi-Pyrenées : Toulouse |
Responsable : Pr Caron, CHU de Toulouse - hôpital Larrey
Pas de centre associé